
Calvin London (calvin@thecomplianceconcierge.com) is the Founder and Principal Consultant of The Compliance Concierge.
Risk management plans (RMPs) for pharmaceutical products originated as regulatory responses to public concerns that pharmaceutical medications were being approved too early. As a result, known problems from using the medication did not become apparent until after introduction into the marketplace.
RMPs are not designed to mitigate adverse events or their reporting. Rather, they are required by some regulatory agencies to specifically manage known risks associated with the use of a drug. Although these go by different names and require differences in design, approval, and operation, they all have a common core element—compliance.
In all medication regimes, there is an unspoken requirement for compliance. While this is often assumed to only be relevant to the manufacturer (also known as the sponsor), RMPs provide an opportunity to look at the broader requirements for compliance at all steps of the process, from drug registration through to patient use. The operation of an RMP is a shared responsibility among the regulatory body, the manufacturer, prescriber, pharmacist, and the patient. This can be viewed as a chain of responsibility, as shown in Figure 1.
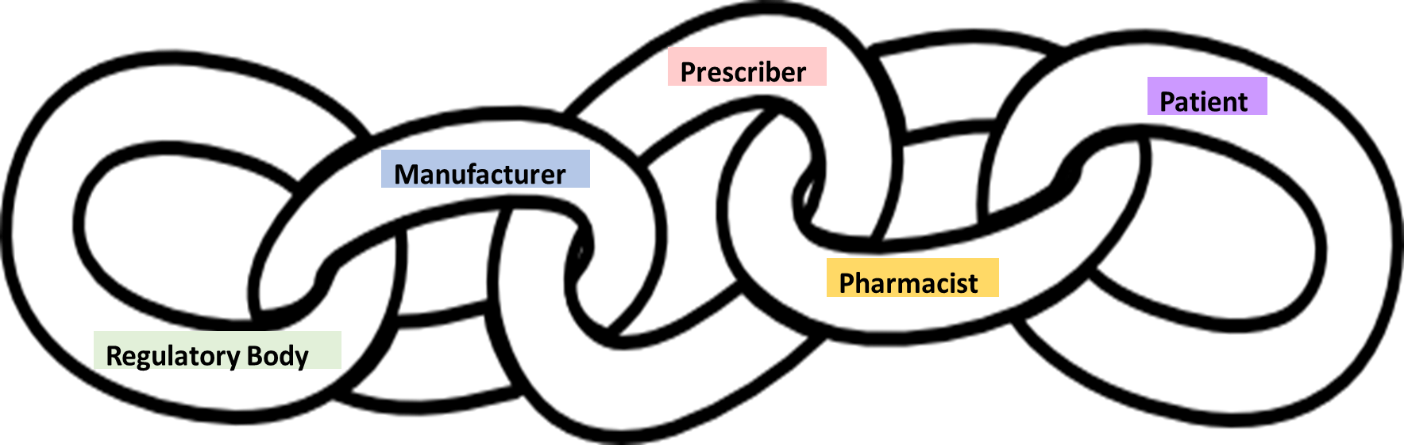
Prescribers and pharmacists both uphold the principles of the RMP. However, as described later, the overall success of the RMP is just as dependent on the individual patient’s sense of responsibility to be compliant. A breakdown in compliance in any of the links—manufacturer, prescriber, pharmacist, or patient—can have significant effects on the effectiveness of the RMP.
Regulatory body
The regulatory body mandates the RMP requirements that the manufacturer must satisfy to maintain compliance with their product registration. In most situations, once this is done, the regulatory body then assumes the manufacturer will execute the RMP and be held responsible for its compliance. As the first link in the chain, they do not have a compliance responsibility as such but do have high expectations of the compliance of the other links to uphold the principles of patient safety.
Manufacturer/sponsor
In the United States, a Risk Evaluation and Mitigation Strategy (REMS) is required by the U.S. Food and Drug Administration for certain medications with serious safety concerns to help ensure the benefits of the medication outweigh its risks.[1] This is the US version of an RMP, and currently, there are some 63 approved REMS.[2] Programs with a similar intent are also required in other regulated markets. For example, the European Medicines Agency requires RMPs for marketing authorizations,[3] as does the Therapeutic Goods Administration in Australia.[4]
In most cases, the nature and extent of the RMP will depend on a combination of special requirements such as testing prior to dispense or use; restricted access to the drug; education of prescribers, pharmacists, and/or patients; and ongoing monitoring of compliance.
Clozapine, for example, which is indicated for the treatment of severely ill patients with schizophrenia who fail to respond adequately to standard antipsychotic treatment, has caused severe neutropenia (defined as an absolute neutrophil count of less than 500/μL).[5] Severe neutropenia can lead to serious infection and death. As such, the RMP for Clozapine products requires the establishment of a minimum level of neutropenic activity prior to use and baseline monitoring during use.[6] Companies that market clozapine provide a database for the monitoring of these results and information related to the use of the drug for physicians to educate patients.
Other examples, and perhaps one of the best known RMPs among the pharmaceutical community, are for the use of Isotretinoin, indicated for the treatment of severe recalcitrant nodular acne, and the immunomodulatory drugs—thalidomide, lenalidomide, and pomalidomide—indicated primarily for the treatment of multiple myeloma. These are not only the most detailed and exacting RMPs, but were also made for the drugs that lead to the global use of RMPs. Readers may be familiar with the issues that resulted from the use of Thalidomide in the 1950s, when the drug was first marketed for morning sickness.[7] Enhanced regulatory compliance, particularly in Australia, Canada, and the United States, was implemented to prevent the issues associated with the development of fetal abnormalities, including amelia (congenital absence of limbs), when Thalidomide was brought back onto the market.[8]
Immunomodulatory drug RMPs call for three areas of manufacturer or drug sponsor compliance: (i) controlled access to the product; (ii) educating prescribers, pharmacists, and patients; and (iii) ongoing compliance monitoring.[9] In short, the requirements of these RMPs include:
-
Registration of prescribers, pharmacists, and patients into a formal program with appropriate consents;
-
Extensive education of prescribers, pharmacists, and patients to qualify for prescribing, dispensing, or using of the drug;
-
Limited supply of drug product to patients, especially those that are considered high risk, as well as women of child-bearing potential (WCBP);
-
Monthly pregnancy testing of WCBP to ensure they are not pregnant while undergoing treatment; and
-
Use of approved contraception by all patients while undergoing treatment.
Additional information for those interested can be found in different reviews.[10]
The compliance responsibility of the sponsor is to establish and maintain a robust RMP and to monitor its performance. As discussed in later sections, the sponsor cannot take total responsibility for the compliance (or lack of) for the other links in the chain. They do, however, need to encase their RMP in a suite of policies and procedures that account for and deal with noncompliance when it occurs.