M.A. Hall (mhall@dugganbertsch.com) is the Healthcare Practice Co-Lead at Duggan Bertsch LLC in Chicago.
Erika Stevens (erika@recherchetransformationrapide.com) is a Principal at Recherche Transformation Rapide in Lumberton, NJ.
Mary Veazie (mlveazie@mdanderson.org) is the Executive Director of Clinical Research Finance at The University of Texas MD Anderson Cancer Center in Houston.
The field of clinical research is no different than any other in the healthcare industry, in that there are federal and state regulations and laws that affect its revenue cycle. Institutions conducting research on humans must comply with all applicable laws and regulations throughout the clinical research cycle in order to continue to provide care for patients, bill for services, and maintain funding. Regulatory requirements and laws impact each phase of the clinical research revenue cycle (CRRC):
-
Institutional review board (IRB)
-
Pre-award, study start-up
-
Post-award
-
Close-out
For this article, we will discuss the compliance risks associated with the regulatory requirements within the pre-award, post-award, and close-out phases of the CRRC and how noncompliance with regulatory requirements across the revenue cycle can affect a clinical research department’s fiscal bottom line. In FY 2017, the federal government recovered $2.4 billion from healthcare fraud actions, which is a return on investment of $6.10 for each dollar spent.[1] For clinical research, understanding compliance risks is essential for everyone, including executive management (C-suite), to avoid significant penalties from federal and state governments. We will discuss and identify the benefits of introducing the concept of proactive compliance to a CRRC program as the most advantageous way of addressing regulatory matters for an institution.
Proactive compliance
Proactive compliance is the system of determining and incorporating the principles of compliance to every process of development or redesign for a CRRC program. New policies, processes, and technologies are only introduced into the revenue cycle of a clinical research program when there is an analysis of the compliance implications. Additionally, to confirm proactive compliance principles were followed by all involved in the CRRC program, there are methods of oversight that allow follow-up to ensure that the concept is recognized and accepted for each CRRC phase that involved regulation. The healthcare fraud laws that apply to the traditional care model are applicable to clinical research, including the False Claims Act (FCA),[2] the Anti-Kickback Statute (AKS),[3] and Stark Law (42 U.S.C. § 1395nn).
Additionally, anti-fraud principles under the FCA that transfer to clinical research include medical necessity, billing for services provided, and unbundling of charges.
These principles put a clinical research program at risk if the financial and study teams running a program are not aware of them or fail to realize the appropriate application. A clinical research department that adopts the proactive compliance approach in the pre-award, post-award, and close-out phases of the CRRC can expect that regulatory concepts that dictate day-to-day operations are considered. This leads to timely identification of risks and results in prioritization throughout the life cycle of a clinical trial or study through the development of controls and other mechanisms.
In addition to the specific liabilities discussed below, a finding of liability against an entity or individual under the AKS, the FCA, or Stark Law can lead to the exclusion from participating as an entity or provider of any services or items reimbursable by federal healthcare programs for periods that can range from three years to permanently. This includes clinical research programs that bill or receive funds from the federal government. Primary authority for the administration, audits of compliance, and enforcement of the federal anti-fraud laws rests with the Office of Inspector General (OIG) of the U.S. Department of Health and Human Services. For criminal prosecutions (when permitted) upon referral by the OIG, the U.S. Department of Justice has primary authority.
Anti-Kickback Statute
The AKS prohibits the exchange of anything of value―cash or “in-kind”―to induce referrals for services or items payable by a federal health program, and the AKS carries penalties of up to $25,000 or imprisonment of not less than 5 years. Under the AKS, the substance of conduct in operating a clinical research program will trump form, that is, the formal language of agreements entered into for purposes of establishing the liability of a party. Targets of enforcement actions, in the cases of entities involved in prohibited conduct, will include the individuals in control of the entities and facility employees.
False Claims Act
The FCA is a civil liability statute that makes it illegal for any person to knowingly make a false record or file a false claim for reimbursement to any federal healthcare program. The primary civil remedies for a violation of the FCA are: (1) imposition of substantial civil monetary penalties, ranging from approximately $10,000 to $21,000 per false claim (meaning per unit of service or item sold for which reimbursement was sought), plus (2) treble damages on the aggregate amount originally paid by the government on the false claim. Under the FCA, the mere filing of a claim for services, the submission of a prescription, or certification of a medical device can be deemed the filing of a false claim in violation of the statute.
Stark Law
The Stark Law (aka the physician self-referral law) prohibits physicians from referring patients for receipt of a whole range of services, commonly referred to as designated health services, if they are to be payable by federal healthcare programs and rendered by entities with which the prescribing physician or members of their immediate family have a direct or indirect financial interest.
The Stark Law is a strict liability statute, that is, specific intent to violate the law is not required for liability to attach, because the violation occurs by the mere referral to an entity where a financial interest is held. Financial interests prohibited by Stark include ownership and investment interests in entities, as well as compensation arrangements involving unaffiliated entities. Additionally, under Stark a physician is prohibited from presenting or causing to be presented claims to a federal healthcare program (or billing another individual, entity, or third-party payer) for the referred services.
Penalties for Stark Law violations include fines up to $15,000 for each billed service, plus three times the amount of the claim overpaid and the possibility of exclusion from participation in federal healthcare programs. Like the AKS, Stark does provide for exemptions (i.e., safe harbors) that permit the holding of such financial interests if structured in accordance with existing regulations.
Each of the regulations and anti-fraud principles detailed here apply to clinical research programs. If an institution is not performing proactively throughout the revenue cycle, it can find itself running afoul of the applicable compliance laws and, thus, having to report incorrect billing and/or return funds along with the associated penalties.
The clinical research revenue cycle
CRRC identifies the activities related to clinical research that affect financial outcomes. Specifically, the revenue cycle captures details for identifying opportunities for revenue, pitfalls for losses, and risks associated with issues of noncompliance in meeting research finance regulatory requirements.[4]
The ability to capture clinical procedures related to research mitigates CRRC compliance risks. Understanding the complexities of compliance risks associated with clinical research is complicated for many organizations, because the potential pitfalls exist throughout the cycle. Financial errors occur in the budgeting, patient identification, charge capture, billing, accounts receivable, and reconciliation processes. Establishing clearly defined processes for review of study-related and non–study-related charges prior to study initiation thwarts future billing issues. Assessing financial feasibility and implementing risk tolerance steps can reduce compliance findings.
In assessing risks, identifying the potential areas of financial/compliance risk throughout the CRRC will enable better safeguards (see Figure 1). For example, in the pre-award phase of the CRRC, many institutions lack a formal clinical research financial budget process, which places the institution at risk of not capturing and recouping all research costs. Further, without a documented coverage analysis, institutions are at risk of potentially billing Medicare for non-qualifying clinical trials and/or potentially inaccurate clinical research billing. In addition, this step is key to the successful execution of the remaining steps in the billing compliance continuum. Institutions may also be losing revenue due to billing denials by a third party for services billable to a sponsor.
During the post-award phase, patient tracking/enrollment registration risks arise without a centralized location for recording all enrolled clinical research participants. Without a central repository, registration and scheduling of these participants is difficult and could lead to errors.
The coding and billing of clinical research activities is complex, and risk occurs without appropriate routing of standard of care vs. research-specific charges. Institutions are at risk for improper billing, loss of revenue due to payment denials, and noncompliance with Medicare clinical research coding and billing regulations.
Another operational risk develops due to inconsistent invoicing and tracking processes that may result in unbilled invoices and loss of potential revenue, such as those related to a clinical trial sponsor. In the absence of established processes and controls, these steps are often overlooked.
Finally, reconciliation risks occur without a periodic reconciliation process, such as loss of revenue and not remediating improper billing in a timely manner.
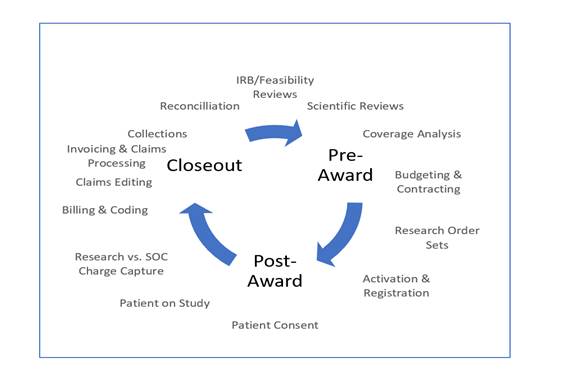
The fiscal regulatory compliance landscape
The pre-award, post-award, and close-out phases of a clinical trial all have their inherent compliance risks.
Pre-award phase (initial regulatory risks)
Optimizing proactive compliance of the CRRC is essential to reducing charge/billing errors and regulatory violations in the pre-award phase. Additionally, verifying that staff have knowledge of relevant regulations and guidance that affect the CRRC in the pre-award phase will significantly reduce the volume of bill holds and refunds that result from incorrect documents and charges. However, this knowledge of the regulations and the compliance policies that dictate CRRC operations must be “baked-in” to the process.
Moreover, a CRRC compliance program is different than other types of healthcare stakeholder compliance programs. For example, federal and private payer billing rules, sponsor contracts, and government regulations all apply, in addition to the different scope in practice.[5] The Centers for Medicare & Medicaid (CMS) Clinical Trial Policy serves as the industry standard for clinical research billing and compliance requirements.[6] In the CMS Medicare Clinical Trial Policy,[7] Medicare coverage includes reimbursements for items and healthcare services that meet the standards for “routine care” (i.e., standard of care) during a clinical trial.
The pre-award processing of coverage analysis is crucial to the CRRC proactive compliance concept. For the CRRC, this is an essential element that must occur without error. During this process, the assigned clinical research finance staff assesses clinical trial documents, practice guidelines, Local Coverage Determinations, and National Coverage Determinations to evaluate the insurance coverage of the study protocol. The Compliance by Design product of the coverage analysis is a detailed billing summary that catalogs what in a study can be billed to insurance, based on regulations, and what the sponsor will reimburse per the study contract. Failing to take the Compliance by Design approach when first developing a preliminary coverage analysis (PCA) or coverage analysis will likely result in delays in the study start-up and significant rework for the clinical research team.
Additionally, deficiencies in the application of regulatory compliance principles to this crucial stage may affect the accuracy and effectiveness of the budgets and study calendars. Finally, a coverage analysis or PCA not drafted in a compliance environment may result in billing errors to the sponsor or insurer that can turn into costly repayment/overpayment issues.
Post-award phase (CRRC compliance risks)
In the post-award phase of the CRRC, compliance risks occur in several areas. Complete and accurate charging/billing for clinical research and standard-of-care procedures requires research subject registration, establishing timely invoicing to sponsors, accurate billing for standard-of-care charges, and identification of Medicare-allowable charges.
Optimizing CRRC processes to reduce charge/billing times will reduce errors and associated delays in rework/reviews, will decrease the volume of refunds stemming from incorrect clinical charges, and will lower the volume of service charges/billing that require reconciliations.
Developing controls on the front, middle, and end of the CRRC process enables proper procedure scheduling, better patient care, and appropriate charging/billing. Finally, streamlining and automating processes to eliminate rework and delay times will further mitigate compliance risks.
Close-out phase (post-reimbursement CRRC compliance risks)
In the close-out phase of the CRRC, a compliance issue repeatedly noted during assessments is that organizations fail to standardize the process for developing a reconciliation plan between services provided, billed, and allowed per the study protocol. As a result, clinical research finance teams are burdened by having to conduct rework between departments to gather solid data regarding the billing and invoicing plan for a study, which is essential for compliance. Finally, lack of standardization in reconciliation of reimbursement sources demonstrates the level of inconsistency between the methods used to develop budgets and increases the likelihood of compliance errors.
Leverage data analytics
Organizations can use data analytics from terminated trials to make various decisions about risks associated with their clinical research portfolio. They can identify trends regarding the proper financial management of trials and use the data on payment terms and on-time payment trends to decide whether to work with particular trial sponsors.
We have some suggestions for developing metrics to help an organization become savvier as it operationalizes resources, developing strategies, and mitigating the financial risks associated with a clinical research portfolio. We also identify important metrics that can help sustain and grow a profitable clinical research program. Key metrics to track and trend are as follows.
Trials submitted to the IRB
To help forecast workforce capacity, examine the number of trials submitted to the IRB each year. Pair down this number to the number of trials that require a Medicare Coverage Analysis (MCA). That total will enable an organization to validate and plan for the staffing needs dedicated to creating the MCA document. The significance of performing this data analysis is twofold.
First, organizations can evaluate whether current staffing plans are sufficient to meet the organization’s goals for trial activation time. However, an additional metric would be to review the length of time necessary for MCA preparation in relation to the activation of a trial. If the activation of a trial was delayed due to the time required for the MCA document preparation, then it’s important to review MCA staffing resources and efficiencies.
Second, the data results enable the organization to develop a high-level overview of the number of trials that represent research clinical care for its patient population.
Different classifications
A comparison of the number of different classifications of trials (i.e., number of sponsor-initiated trials vs. number of investigator-initiated trials vs. number of federally sponsored trials) spotlights the existing research program’s mix. Each type of sponsor has various funding expectations. Federally sponsored and investigator-sponsored trials have lower funding, whereas external sponsors (e.g., pharmaceutical companies) provide higher funding. Without external sponsors, the financial risk increases, because the organization will have to use more of its own resources to fund the trial. Monitoring the complement of sponsored trials helps ensure that the organization maintains its desired mix of trials and guides associated financial strategies that support research.
Trials by therapeutic area
Evaluation of this metric can help determine whether the portfolio is aligned with an organization’s patient population. A clinical research program whose trials have difficulty enrolling subjects due to a low patient population for the trial’s therapeutic area have a negative impact on revenue. Additionally, costs for studies that involve certain therapeutic areas will be higher than trials that involve other therapeutic areas. This metric, coupled with a review of the average-cost-per-participant metric, will shed light on the profitability associated with the trial portfolio.
Cost per participant
The average cost per patient is derived by dividing the total cost of conducting the trial by the number of enrolled participants, regardless of whether those participants completed the entire study. The resulting data yields more than just the expense associated with the trial; it can also point to a type of a trial that may be extremely costly to conduct and that had difficulties enrolling participants.
Cost to start trial
Administrative costs associated with activating a trial can be far greater than expected if the organization doesn’t review the average cost to start a trial. Armed with this information, the organization may recognize the need to examine operational resources supporting activation and look for ways to increase efficiencies with the goal of cost reduction.
Trial submission to activation
One of the most significant metrics for an organization that is hoping to attract external sponsors with scientifically valuable trials is the speed with which a principal investigator (PI) can activate the trial for new participant enrollment. The longer it takes to open a trial, the costlier the study becomes, because patients cannot enroll and the milestone payments from enrollment and clinical events are not received.
Long-term trends
The impact on an organization’s clinical research portfolio is even more significant in the long term. Historical activation trends are among the most critical of a sponsor’s assessments when determining the feasibility of sponsoring a trial with an organization.
First participant on trial
This data point represents the time it takes for the first study participant to enroll in the trial. This measure is significant, because it reflects the PI’s ability to recruit and select trials that work for the organization’s patient population. It is also important for some of the same reasons that the average-time-from-trial-submission-to-activation metric is important: Delay in enrollment is costly and sends a negative message about the PI’s commitment to a clinical study.
Insurance denials per trial
Data results from this metric will require further investigation if there is a higher denial rate than expected. Any one or more of the following elements associated with the trial may have contributed to increased denials:
-
Accuracy of the MCA: If the denials data demonstrates a trend over time for a disease and/or procedure, then the organization should carefully evaluate the determinations denoted on the MCA. Information gained from this evaluation will assist with future MCA developments on the next selection of trials.
-
Accuracy of the ordering specifications: Understanding the intent of ordering the item or service helps downstream workflow, which enables more accurate and appropriate processing. If frequent errors are discovered due to how an item or service was ordered, the organization can correct those errors.
-
Medical documentation accuracy: Insurance reimbursements rely heavily on supporting accurate medical documentation. In clinical research care, the need for accurate documentation becomes critical, because insurance companies are reluctant to reimburse for research-related procedures.
-
Trial therapeutic area: Trials for certain disease categories have higher expenses than those for other therapeutic areas. A more in-depth examination of the types of insurance denials in a given disease category for a particular item or service will help the PI and the organization determine the financial feasibility of conducting similar trials in the future.
Revenue collected per sponsor
This data can provide organizations with targeted collection capabilities related to a sponsor. It can help organizations decide to continue or discontinue business with that sponsor.
Profit and loss per trial
The profit-and-loss data and residual funds data can provide valuable information about the strength of the organization’s trial portfolio, but it also may indicate a weakness in operational activities supporting clinical research. For example, budgets and contracts alone may be insufficient to capture the true costs of the study.
Conclusion
Evaluating the actual cost and risk of conducting clinical research is a complicated endeavor for any organization. Multiple metrics must be taken into consideration. It is often challenging for an organization to distinguish between medical care expenses and research medical care expenses, because a patient will often incur both while participating in a trial. Committing to invest in data analytics and then using the data as a resource will allow an organization to make the informed strategic decisions necessary to manage a successful clinical research portfolio.
Takeaways
-
A methodical compliance approach in clinical research is essential to risk mitigation.
-
Application of compliance principles throughout the clinical research process enables CCRC compliance.
-
State and federal regulatory issues apply to clinical research revenue cycle.
-
Not having a robust clinical research compliance program throughout the revenue cycle can affect the bottom line and the ability of an institution to take on further studies.
-
Clinical research revenue cycle compliance begins from the initial start of the study process.
-